TL;DR:
- Assay labs are central to product development, supporting quality, safety, and regulatory compliance.
- Selecting validated, stage-appropriate methods and integrating testing early mitigates risks and delays.
- Using assay data proactively drives innovation, reduces costs, and streamlines regulatory approval.
Most product development teams treat assay labs as a late-stage formality. Submit samples, wait for a green light, move on. That framing is costly. Assay labs are where your material’s identity, purity, potency, and stability are confirmed or challenged, and the data they generate shapes regulatory submissions, shelf-life claims, and go-to-market timelines. For biomedical and pharmaceutical teams, the difference between a lab used reactively and one used strategically can mean months of delay or a clean agency review. This guide covers what assay labs actually do, which methodologies matter most, how regulatory frameworks have evolved, and how to turn testing from a compliance checkbox into a genuine competitive advantage.
Table of Contents
- What are assay labs and why do they matter?
- Key testing methodologies in assay labs
- Regulatory requirements and validation: What product teams must know
- Beyond compliance: How assay labs drive innovation and savings
- Field perspective: What most teams miss about assay labs
- Advanced solutions for your assay and materials testing needs
- Frequently asked questions
Key Takeaways
| Point | Details |
|---|---|
| Assay labs’ vital role | Assay labs are essential for both regulatory compliance and product innovation in pharma and biomed sectors. |
| Method diversity | Advanced labs use a range of techniques like DSC, HPLC, and GC-MS for purity, potency, and stability. |
| Compliance evolution | Modern validation demands lifecycle thinking and data-driven decision-making, not just traditional ‘three-batch’ tests. |
| Innovation advantage | Proactive lab use drives faster development, cost savings, and risk reduction beyond regulatory requirements. |
| Partnering for success | Engaging with full-service labs ensures readiness for regulatory changes and complex material challenges. |
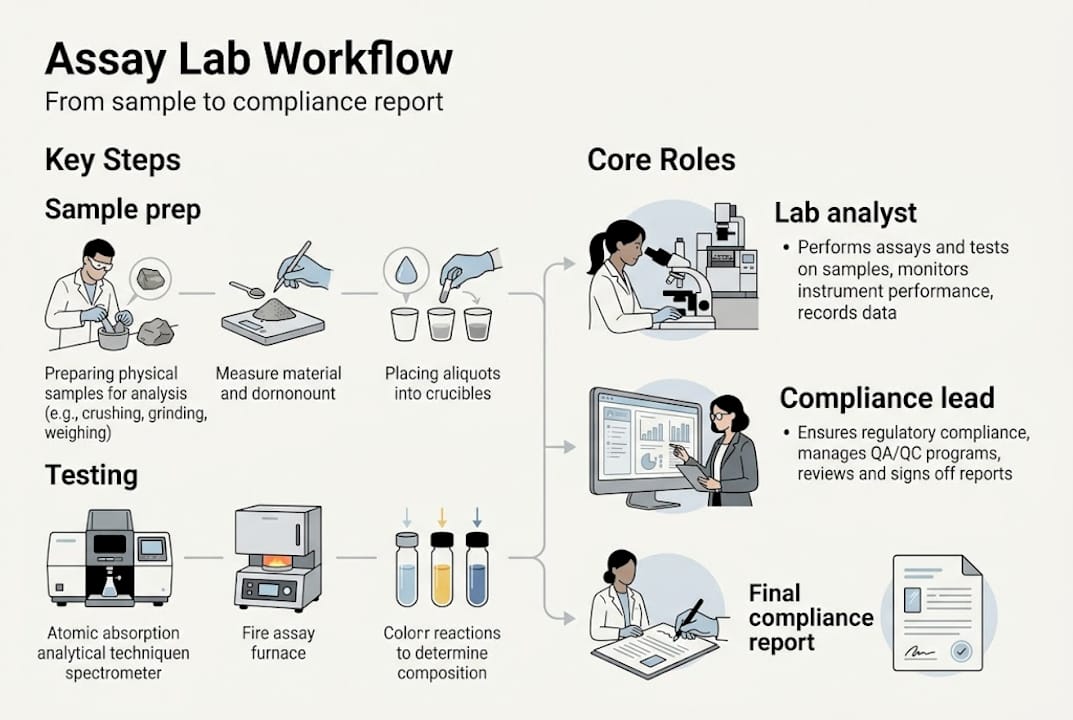
What are assay labs and why do they matter?
An assay lab is not simply a place where samples are checked against a pass/fail threshold. Assay labs conduct quantitative measurement of biological or chemical properties using techniques like HPLC, GC-MS, ELISA, DSC, and TGA to evaluate purity, potency, stability, and material composition. That range of capability positions them at the center of product development, not at its end.
In practice, assay labs support material characterization across the full development lifecycle. Early-stage work might focus on identifying impurity profiles or confirming raw material identity. Later stages shift toward stability studies, potency verification, and batch release testing. Each phase generates data that either supports or complicates your regulatory package.
The importance of analytical testing becomes clear when you consider what agencies actually require. FDA, ICH, CAP, and CLIA frameworks all depend on validated assay data to confirm that a product performs as labeled. A common regulatory target is 95 to 105% label claim for potency, and missing that window without documented justification triggers queries, holds, or worse.
The misconception that assay labs exist only for regulatory sign-off creates real costs. Teams that wait until submission preparation to run full assay panels often discover out-of-spec results that require root cause investigation, repeat testing, and timeline extensions. Those delays are preventable when assay data is integrated earlier.
Key functions of modern assay labs include:
- Regulatory compliance support: Generating validated data aligned with FDA, ICH, CAP, and CLIA requirements
- Innovation enablement: Providing material performance data that informs formulation decisions and design iterations
- Cost reduction: Identifying impurity or stability issues before scale-up, when corrections are far less expensive
- Risk mitigation: Flagging out-of-spec trends during development rather than during batch release or post-market
- Shelf-life and stability support: Producing the empirical data needed for expiry dating and storage condition claims
Thinking of your assay lab as an extension of your development team, rather than a downstream checkpoint, changes how and when you use it.
Key testing methodologies in assay labs
Choosing the right analytical method is not a formality. The technique must match the material, the regulatory context, and the stage of development. Here is how the core methodologies break down:
| Technique | What it measures | Key application |
|---|---|---|
| DSC (Differential Scanning Calorimetry) | Thermal transitions, melting points, crystallinity | Polymorphism, formulation stability |
| TGA (Thermogravimetric Analysis) | Mass loss with temperature | Decomposition, moisture content |
| FTIR / Raman / XPS | Molecular bonds, surface chemistry | Composition ID, contamination screening |
| HPLC / GC-MS | Purity, impurity profiling, potency | Drug content, residual solvents |
| Tensile / Hardness Testing | Mechanical strength, tablet hardness | Device integrity, dosage form performance |
Thermal stability testing via DSC and TGA is especially valuable for pharmaceutical materials because it reveals how a compound or excipient behaves under temperature stress, which directly informs packaging and storage specifications. Spectroscopy methods like FTIR and Raman are used for rapid composition verification and contamination screening, while XPS provides surface-specific elemental data critical for device coatings and implant materials.

For solid dosage forms and medical devices, mechanical testing is often underutilized. A chemical analysis overview of a tablet formulation is incomplete without knowing its hardness or friability. Tensile and compression testing reveals how a device or dosage form will perform under real-world stress, and basic chemical assays like gravimetric analysis confirm mass-based purity with high precision.
Method selection should also align with ICH Q1A stability guidance and FDA method validation requirements. A method validated for a clinical batch may need re-validation if the manufacturing process changes.
Pro Tip: Match your method to your development stage. Early R&D benefits from broad screening techniques like GC-MS and FTIR. Clinical and commercial batches require fully validated, compendial-aligned methods with documented precision, accuracy, and specificity.
Regulatory requirements and validation: What product teams must know
Regulatory expectations for assay validation have shifted significantly. The traditional “3-batch” model, where three consecutive batches were used to validate a process, has given way to FDA’s 3-stage lifecycle approach: process design, process qualification, and continued process verification. This is not a minor procedural update. It requires teams to think about validation as an ongoing commitment, not a one-time event.
The number of batches used for validation is no longer fixed. Statistical justification is required, meaning your team must demonstrate that the chosen sample size provides meaningful confidence in process consistency. Arbitrary batch counts no longer satisfy agency reviewers.
For potency and stability, ICH Q1A sets the standard conditions: 25°C/60% RH for long-term studies and 40°C/75% RH for accelerated testing. Shelf-life claims must be supported by data from these conditions, and potency results must fall within the 95 to 105% label claim window throughout the claimed shelf life.
Steps for regulatory-compliant assay validation:
- Define the intended purpose and regulatory context for each method
- Select compendial or custom methods based on material type and submission requirements
- Establish acceptance criteria grounded in statistical analysis, not convention
- Execute validation runs with documented precision, accuracy, specificity, and linearity
- Generate a validation report aligned with FDA and ICH Q2(R1) guidance
- Implement continued verification protocols to monitor method performance post-approval
In-house validation offers control and speed for teams with established infrastructure. Outsourcing to a contract research organization (CRO) or specialized lab provides scalability and access to validated methods without capital investment. The trade-off is coordination overhead and the need for robust data transfer protocols.
Method validation strategies and batch consistency evaluations are two areas where partnering with a specialized lab pays dividends, particularly when your internal team is managing multiple programs simultaneously.
Beyond compliance: How assay labs drive innovation and savings
The financial case for strategic assay lab use is well documented. Orthex reduced testing costs by 20% by optimizing their lab network selection, demonstrating that smart sourcing of analytical services is itself a cost-containment strategy. For most biomedical and pharmaceutical teams, testing spend is not fixed. It scales with how well you plan.
Robust assay data also accelerates go-to-market timelines. Shelf-life prediction using Arrhenius modeling, which extrapolates long-term stability from accelerated testing data, allows teams to establish expiry claims before real-time studies are complete. That is not a shortcut. It is a scientifically validated approach that agencies accept when the underlying data is sound.
Ways to leverage assay labs for strategic advantage:
- Early impurity profiling: Identify degradation pathways before scale-up, when formulation changes are still low-cost
- Forced degradation studies: Stress-test your material under heat, light, humidity, and pH extremes to map its vulnerability profile
- Extractables and leachables (E&L) testing: Critical for device and combination products; often overlooked until late in development
- Shelf-life modeling: Use accelerated stability data to support provisional expiry claims and reduce real-time study timelines
- Specification setting: Use assay data to set limits that reflect actual product performance, not inherited defaults
“Data-driven specifications over arbitrary limits” is not just a best practice. It is the standard regulators now expect, and teams that set specs based on real assay data face fewer agency challenges during review.
Stage 3 continued verification is where most teams fall short. Post-approval monitoring of assay performance catches method drift, raw material variability, and process shifts before they become compliance events. Custom materials testing programs built around Stage 3 requirements give your team the early warning system that prevents costly investigations.
Field perspective: What most teams miss about assay labs
We see a consistent pattern across product development teams: assay labs get engaged when a regulatory trigger appears, not before. The FDA submission is approaching, the QA team flags a gap, and suddenly the lab is asked to run a full validation package under time pressure. That sequence is backwards.
Elite teams use assay data to manage risk proactively. They run forced degradation studies during formulation development to understand what their product is capable of under stress. They conduct E&L assessments for device components before design lock. They build Stage 3 verification into their commercial quality systems from day one, not as an afterthought.
The teams that avoid FDA 483 observations are not the ones with the most resources. They are the ones who treat their assay lab as a decision-support tool throughout development. Out-of-spec investigations, specification justifications, and stability failures are far less disruptive when the underlying data infrastructure is already in place.
Our perspective is straightforward: an analytical testing mindset that integrates assay data early transforms compliance from a reactive scramble into a managed process. Labs are not regulatory hurdles. Used correctly, they are the clearest signal you have about whether your product is ready.
Advanced solutions for your assay and materials testing needs
If your team is ready to move beyond reactive testing, we are here to support that transition. At Materials Metric, our analytical testing methods are designed to align with FDA, ICH, and GLP/GMP requirements from the first sample submission.
We provide elemental characterization services alongside spectroscopy, thermal analysis, mechanical testing, and biocompatibility evaluation, giving your team a single point of contact for complex, multi-method programs. Our advanced material characterization capabilities are built to support both early-stage R&D and commercial compliance needs. Reach out to discuss how we can become an extension of your development team.
Frequently asked questions
How do I select the right assay lab for my project?
Match your project to a lab with validated methods and regulatory expertise for your specific material type and submission pathway. Capability breadth matters less than demonstrated experience with your testing requirements.
What is the typical turnaround time for advanced materials testing at assay labs?
Routine assays typically complete in one to three weeks, while custom method development and full validation packages can take several months depending on regulatory complexity and sample volume.
How can assay labs help with FDA or ICH compliance?
Assay labs generate validated, reproducible data for potency, stability, and impurity profiling aligned with CFR, FDA, and ICH guidance, providing the documented evidence base needed for audit readiness and submission support.
What common mistakes cause assay failures or compliance issues?
Skipping Stage 3 continued verification and setting specifications based on convention rather than empirical data are the two most common drivers of FDA 483 observations and out-of-spec investigation backlogs.


