Most medical device teams treat the 510(k) process as a paperwork problem. Gather the testing data, fill in the FDA templates, submit, and wait. But that assumption is where many companies lose months and significant budget. The FDA 510(k) pathway requires demonstrating substantial equivalence to a legally marketed predicate device through premarket notification, and the details of that demonstration are where submissions succeed or collapse. Shifting reviewer expectations, evolving cybersecurity guidance, and strict documentation standards mean that even experienced regulatory teams face holds and additional information requests on submissions they believed were complete.
Table of Contents
- What is 510(k) and who needs support?
- Key steps and requirements in the 510(k) process
- Common pitfalls and how support stops costly mistakes
- Comparing support options: DIY vs. consultant-led 510(k)
- Essential tests and documentation for 510(k) success
- Why 510(k) support is more than regulatory ‘box checking’
- Ready for streamlined, compliant 510(k) support?
- Frequently asked questions
Key Takeaways
| Point | Details |
|---|---|
| Substantial equivalence is critical | 510(k) support helps ensure your device matches FDA requirements for predicate comparison. |
| Common pitfalls increase delays | Incomplete testing and poor predicate selection are top sources of costly 510(k) hold-ups. |
| Expert consulting accelerates approval | Strategic support reduces rework, RTA/AI rates, and helps you navigate regulatory edge cases. |
| Robust documentation ensures compliance | Comprehensive technical data—especially for biocompatibility and cybersecurity—is mandatory for successful FDA clearance. |
What is 510(k) and who needs support?
The 510(k) premarket notification is the FDA’s mechanism for evaluating medical devices before they reach the market. Its core purpose is to establish that a new device is substantially equivalent to a predicate, meaning it shares the same intended use and has the same or equivalent technological characteristics. When technological differences exist, the manufacturer must demonstrate that those differences do not raise new safety or effectiveness questions.
The pathway applies primarily to Class II devices, which represent the largest category of medical devices cleared in the United States. Class I devices often qualify for exemptions, while Class III devices generally require more rigorous premarket approval. Understanding where your device sits within this classification framework is the essential first decision your regulatory team must make.
Who specifically needs to submit a 510(k)?
- Manufacturers introducing a new Class II device to the US market for the first time
- Companies modifying an existing device in ways that could significantly affect safety or effectiveness
- International manufacturers seeking FDA clearance to sell in the US market
- Startups and first-time submitters without an established regulatory infrastructure
- Device companies re-entering the market after a product discontinuation
Why does external support matter so much? Because demonstrating substantial equivalence is not simply a matter of listing similarities. It requires structured evidence, appropriate predicate selection, targeted testing, and documentation that anticipates reviewer questions before they arise. For companies without deep FDA submission experience, each of these requirements represents a potential gap that can trigger a hold or rejection.
Key steps and requirements in the 510(k) process
With a clear understanding of what 510(k) is, let’s walk through the most important steps and requirements that define submission success, including why many companies miss the mark.
A well-structured submission follows a logical sequence that builds from device description through safety and effectiveness evidence to a final clearance determination. Here are the core stages:
- Device classification and predicate selection. Confirm your device’s classification and identify one or more predicates that share intended use and technological characteristics. This step directly affects the rest of your submission.
- Substantial equivalence comparison. Prepare a detailed comparison document showing how your device’s intended use and technological characteristics align with the predicate. Address any differences with supporting data.
- Performance testing and bench testing. Design and execute the required testing based on your device type. This includes mechanical, electrical, and functional performance evaluations.
- Biocompatibility evaluation. Conduct a thorough biocompatibility assessment per ISO 10993, following a biocompatibility preparation checklist aligned with FDA’s current guidance.
- Electromagnetic compatibility (EMC) testing. For electrically powered devices, EMC testing demonstrates the device performs safely in its intended environment without interfering with other equipment.
- Cybersecurity documentation (for SaMD). Software as a Medical Device requires a cybersecurity management plan, threat modeling, and evidence of secure development practices.
- Labeling review. Confirm all labeling meets FDA requirements for clarity, intended use description, contraindications, and warnings.
- Administrative review against the RTA checklist. Before submitting, verify every element against the Refuse to Accept checklist. Missing even one administrative item can delay your submission immediately.
Poor predicate selection is among the most consequential early-stage mistakes. Selecting a recalled or inferior predicate increases device recall risk 6.4 times, which means a flawed predicate choice doesn’t just affect clearance timing. It can shape the device’s entire post-market risk profile. Class III 510(k)s carry additional requirements, including a summary or certification of the information in the submission.
Pro Tip: Before finalizing your predicate, search the FDA’s 510(k) database for any safety communications or recalls associated with that device. A clean predicate history reduces review complexity and long-term liability.
Common pitfalls and how support stops costly mistakes
Next, let’s take a closer look at why so many submissions don’t pass the first time and how working with specialists can dramatically reduce those risks.
The FDA’s own performance data tells a clear story. In 2025, the average 510(k) review time was approximately 146 days, with a median of 142 days for AI/ML devices. About 24% of submissions were cleared in under 90 days, and 67% received an additional information (AI) request during review. A 33% rate of Refuse to Accept (RTA) holds means roughly one in three submissions fails the administrative gate before scientific review even begins.
“The eSTAR mandate reduced RTA holds from 60% to 33%, but that still means a third of submissions face immediate delays at the administrative review stage before substantive scientific evaluation starts.” (FDA 510k Success Rate & Rejection Reasons Guide)
What drives these holds and delays? The patterns are consistent:
- Incomplete RTA checklist items: Missing sections, unsigned declarations, or absent device descriptions trigger immediate holds.
- Insufficient biocompatibility data: FDA reviewers frequently request additional data when manufacturers rely on material comparisons alone rather than direct device-level testing. Our biocompatibility test compliance services are designed to produce submission-ready data from the start.
- Cybersecurity gaps for SaMD: Submissions for software devices without a robust cybersecurity file consistently attract AI requests.
- Poor or risky predicate choices: Using a predicate that has been recalled or that differs significantly in technological characteristics creates a weak foundation that reviewers will challenge.
- Missing or vague labeling content: Unclear indication statements or missing contraindications generate follow-up requests that extend review timelines significantly.
The value of specialist support shows up most clearly at two moments: during submission preparation, when expert review catches gaps before the FDA sees them, and during the AI response phase, when precise, well-structured answers to FDA questions can prevent a second round of information requests.

Pro Tip: When responding to an FDA additional information request, answer only what is asked. Volunteering extra information outside the specific question can inadvertently open new lines of inquiry that extend your review timeline.
Working with experienced regulatory consulting strategies also means your team benefits from pattern recognition built across many submissions. Specialists know which reviewer concerns appear most frequently for specific device types, and they can structure your submission to proactively address those concerns before they generate AI requests.
Comparing support options: DIY vs. consultant-led 510(k)
Understanding the obstacles, it’s critical to assess whether your team should go it alone or bring in seasoned support. Here is a direct comparison.
| Factor | DIY submission | Consultant-led submission |
|---|---|---|
| Initial cost | Lower upfront spend | Higher upfront investment |
| Timeline risk | Higher (33% RTA rate, 67% AI rate) | Reduced through expert preparation |
| Rework cost | High if RTA or AI requests occur | Minimized by anticipating FDA needs |
| Predicate strategy | Risk of poor selection without historical knowledge | Informed by cross-submission experience |
| Documentation quality | Variable, dependent on internal expertise | Structured, reviewer-ready from the start |
| Best suited for | Simple devices with experienced internal teams | First-timers, novel technologies, SaMD, complex devices |
| Post-clearance value | Minimal additional infrastructure | Documentation supports future submissions |
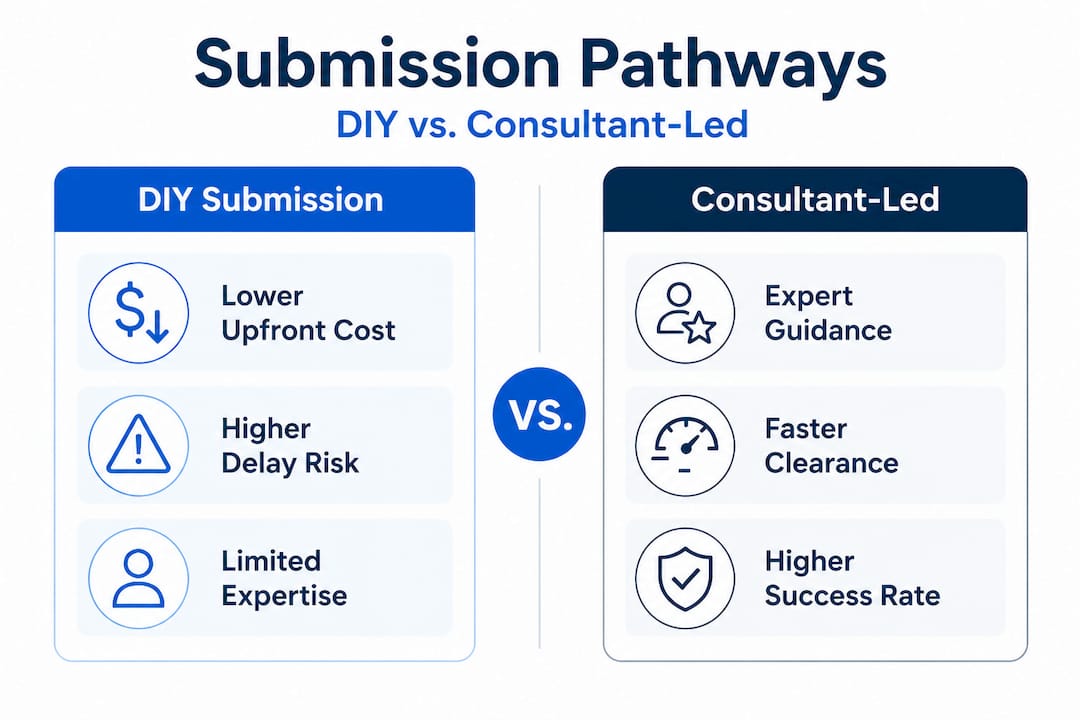
The case for quality consulting for compliance is particularly strong for first-time submitters. Companies without prior 510(k) experience face a steeper learning curve precisely when stakes are highest. They are more likely to encounter RTA holds or AI requests, both of which add weeks or months to a product launch timeline. For a medical device company, a delayed FDA clearance is not just a scheduling inconvenience. It represents delayed revenue, extended development costs, and competitive risk.
The costs of going it alone often exceed the cost of consulting when rework, extended timelines, and additional testing cycles are factored in. Experienced teams know where submissions are most likely to break and they build defenses around those points from the beginning. That structural preparation is the core value of specialist support.
Essential tests and documentation for 510(k) success
Finally, no submission is complete without robust supporting evidence. Let’s explore the essential tests and how to present them effectively.
The testing requirements for a 510(k) depend on device type, materials, intended use, and patient contact. The table below outlines the primary testing categories and their relevance:
| Test category | Applicable to | Key standard or guidance |
|---|---|---|
| Biocompatibility | All patient-contacting devices | ISO 10993 series |
| EMC testing | Electrically powered devices | IEC 60601-1-2 |
| Electrical safety | Active devices | IEC 60601-1 |
| Software validation | All software-containing devices | FDA Software Guidance (2023) |
| Cybersecurity | SaMD and connected devices | FDA Cybersecurity Guidance (2023) |
| Sterility and shelf life | Sterile devices | ISO 11135, ASTM F1980 |
| Mechanical performance | Structural and implantable devices | Device-specific standards |
For software and AI/ML-based devices, the 510(k) pathway imposes requirements that go beyond the hardware device world. A Software as a Medical Device submission must include a Software Bill of Materials (SBOM), a cybersecurity management plan, anomaly resolution records, and evidence of software verification and validation. AI/ML devices face even more scrutiny because their decision logic may evolve over time, requiring predetermined change control plans.
Here is the sequence we recommend for building a complete evidence package:
- Define patient contact and exposure duration to determine the applicable ISO 10993 testing matrix for biocompatibility.
- Identify all applicable consensus standards and prepare a declaration of conformity or deviation justification for each.
- Execute biocompatibility testing at an accredited laboratory, referencing a complete biocompatibility testing guide to ensure every endpoint is addressed.
- Complete EMC and electrical safety testing in an ISO 17025-accredited facility to produce test reports FDA reviewers will accept.
- Prepare the software documentation package including architecture design charts, hazard analysis, and validation test protocols.
- Develop cybersecurity documentation including threat modeling results, penetration testing outcomes where applicable, and update mechanisms.
- Assemble the complete submission package using eSTAR, verifying every RTA checklist item is satisfied before final upload.
The eSTAR electronic submission format has significantly reduced administrative RTA holds, cutting them from 60% to 33% as noted in recent performance data. However, using eSTAR correctly requires understanding how the template maps to the underlying regulatory requirements. Formatting errors within eSTAR can still trigger holds if required sections are blank or improperly structured.
Why 510(k) support is more than regulatory ‘box checking’
Here is the part that rarely appears in regulatory guidance documents or standard consulting proposals: the real value of expert 510(k) support is not in helping you submit the right paperwork. It is in helping you understand whether your product is ready to be a market success before the FDA ever reviews it.
We have seen submissions where the testing data is technically complete but reveals a material biocompatibility concern that would have made the device unmarketable. Without the expert context to interpret those results early, a company might push through the submission, receive clearance, and then discover that their product triggers adverse reactions at a rate that generates post-market surveillance findings. The regulatory strategy insights that matter most are the ones that prevent these scenarios before they happen.
There is also a data legacy argument that most companies miss. The documentation and testing data assembled for a 510(k) is not single-use. It forms the foundation for subsequent submissions for next-generation devices, line extensions, and international regulatory filings. Companies that invest in high-quality, well-organized submission packages build an asset that accelerates future regulatory work. Companies that take shortcuts to get a submission out the door often find themselves starting from scratch on the next product.
For AI/ML device developers, this point is even more critical. The FDA’s evolving posture on algorithm transparency, predetermined change control plans, and post-market performance monitoring means that the regulatory strategy you establish at 510(k) submission sets the terms for your ongoing relationship with FDA oversight. Expert support at this stage is not just about getting cleared. It is about establishing a credible, scalable regulatory foundation for a product that will continue to evolve.
The uncomfortable truth is that many companies underestimate support costs, then overestimate their own internal capabilities, and end up paying more in delays, rework, and additional testing cycles than they would have spent on expert guidance from the beginning. Strategic preparation is not a premium service for well-resourced companies. It is the most cost-effective path for any company serious about getting to market efficiently.
Ready for streamlined, compliant 510(k) support?
Navigating the 510(k) process demands more than regulatory familiarity. It requires precise testing data, strategically structured documentation, and an understanding of how FDA reviewers will interpret every element of your submission.
At Materials Metric, we act as an extension of your regulatory and product development teams. From analytical testing compliance that produces submission-ready data to specialized biocompatibility tests for devices aligned with ISO 10993 and FDA guidance, our services are built to reduce delays and strengthen your submission quality. We combine accredited testing capabilities with regulatory consulting expertise so your team enters the submission process with confidence, not uncertainty. Contact us to discuss your device and build a testing and documentation strategy that gets you to clearance faster.
Frequently asked questions
How long does the FDA 510(k) review process take?
The average review time is approximately 146 days, but 24% of well-prepared submissions are cleared in under 90 days, making thorough upfront preparation the most effective way to accelerate your timeline.
What is the most common reason 510(k) submissions are delayed?
Most delays trace back to incomplete or weak submissions, including missing testing data, poor predicate selection, and insufficient documentation for biocompatibility, EMC, or cybersecurity requirements.
Can software and AI-based medical devices use the 510(k) pathway?
Yes, but AI/ML and SaMD submissions must satisfy additional cybersecurity and validation requirements, and they face a higher rate of additional information requests during review compared to traditional hardware devices.
What is the benefit of 510(k) consulting versus in-house submission?
Consultants reduce the risk of costly RTA holds and AI requests through structured preparation, particularly valuable for first-time submitters and companies developing novel or software-driven medical devices.


